Session 4: Coordinated Multiple Views
Contents
Session 4: Coordinated Multiple Views#
This notebook demonstrates how to create coordinated multi-view visualizations using gos with two examples.
We recommend opening this notebook using Chrome.
You will learn three key features for creating coordinated multi-view visualization:
Composing multiple views (
gos.vertical,gos.horizontal,gos.overlay)Brushing and linking (
linkingId)Linking views (
linkingId)
Start by importing gosling and other required python packages.
!pip install gosling==0.0.9
import gosling as gos
from google.colab import files
import json
Requirement already satisfied: gosling==0.0.9 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (0.0.9)
Requirement already satisfied: pandas in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from gosling==0.0.9) (1.4.3)
Requirement already satisfied: jinja2 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from gosling==0.0.9) (3.1.2)
Requirement already satisfied: jsonschema<4.0,>=3.0 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from gosling==0.0.9) (3.2.0)
Requirement already satisfied: six>=1.11.0 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from jsonschema<4.0,>=3.0->gosling==0.0.9) (1.16.0)
Requirement already satisfied: pyrsistent>=0.14.0 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from jsonschema<4.0,>=3.0->gosling==0.0.9) (0.18.1)
Requirement already satisfied: setuptools in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from jsonschema<4.0,>=3.0->gosling==0.0.9) (58.1.0)
Requirement already satisfied: attrs>=17.4.0 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from jsonschema<4.0,>=3.0->gosling==0.0.9) (21.4.0)
Requirement already satisfied: MarkupSafe>=2.0 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from jinja2->gosling==0.0.9) (2.1.1)
Requirement already satisfied: pytz>=2020.1 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from pandas->gosling==0.0.9) (2022.1)
Requirement already satisfied: numpy>=1.21.0 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from pandas->gosling==0.0.9) (1.23.0)
Requirement already satisfied: python-dateutil>=2.8.1 in /opt/hostedtoolcache/Python/3.10.5/x64/lib/python3.10/site-packages (from pandas->gosling==0.0.9) (2.8.2)
---------------------------------------------------------------------------
ModuleNotFoundError Traceback (most recent call last)
Input In [1], in <cell line: 4>()
1 get_ipython().system('pip install gosling==0.0.9')
2 import gosling as gos
----> 4 from google.colab import files
5 import json
ModuleNotFoundError: No module named 'google'
Example 1: Overview + Detail#
An overview+detail presents 1) a rough overview of the complete information without details and 2) a small portion of the information with details.
A brush is often used to select a subset of data items from the overview.
👉 Create a bar chart#
data = gos.multivec(
url="https://server.gosling-lang.org/api/v1/tileset_info/?d=cistrome-multivec",
row="sample",
column="position",
value="peak",
categories=["sample 1", "sample 2", "sample 3", "sample 4"],
binSize=4,
)
bar_chart = gos.Track(data).mark_bar().encode(
y="peak:Q",
x=gos.X("start:G", axis="bottom", domain=gos.GenomicDomain(chromosome="7")),
xe='end:G',
color=gos.Color("sample:N", legend=True), # add color legend
stroke=gos.value("black"),
strokeWidth=gos.value(0.3),
).properties(height=100, width=800)
bar_chart.view()
👉 Create a detailed view#
We can create a detailed view for the same data by changing the genomic domain (x) of the bar chart we just created.
Apart from focusing on a smaller genomic domain, we also 1) change the chart background, 2) modify the chart height, and 3) split the stacked bar chart for the detailed view.
🇮 Note
Remember to assign a uniquelinkingIdtox(the genomic domain).
This id will be used later to link this detailed view and a brush on the overview.
linkingId = 'brush1'
brushColor = 'steelblue'
detail = bar_chart.encode(
row='sample:N', # split the stacked bar chart
style=gos.Style(background=brushColor, backgroundOpacity=0.2), # change background color
x=gos.X(
"start:G",
axis="bottom",
domain=gos.GenomicDomain(chromosome="7", interval=[77700000, 81000000]),
linkingId=linkingId # this linkingId will be used later to link this detailed view to a brush in the overview
),
).properties(height = 200)
detail.view()
👉 Linking the overview and the detail view through a brush#
# we do not change the bar_chart but create a new object by modifying its mark type
brush_track = bar_chart.mark_brush().encode(
x=gos.X(
"start:G",
linkingId=linkingId # the brush has the same linkingId as the detailed view
),
color=gos.value(brushColor)
)
So far, we already have all the components needed for an overview+detail visualization: bar_chart, brush_track, detail.
Let’s compose them together to get a multi-view visualization!
# create an overview by overlaying the brush upon the bar chart
overview = gos.overlay(
bar_chart,
brush_track
)
# compose the detailed view and the overview
gos.vertical(
detail,
overview
)
👉 Add more detail views#
linkingId2='brush2'
brushColor2='green'
brush_track2 = bar_chart.mark_brush().encode(
x=gos.X(
"start:G",
linkingId=linkingId2
),
color=gos.value(brushColor2)
)
overview_2brush = gos.overlay(
bar_chart,
brush_track,
brush_track2
)
detail2 = detail.encode(
style=gos.Style(background=brushColor2, backgroundOpacity=0.2),
x=gos.X(
"start:G",
axis="bottom",
domain=gos.GenomicDomain(chromosome="7", interval=[47700000, 51000000]),
linkingId=linkingId2
),
)
gos.vertical(
detail,
detail2,
overview_2brush
)
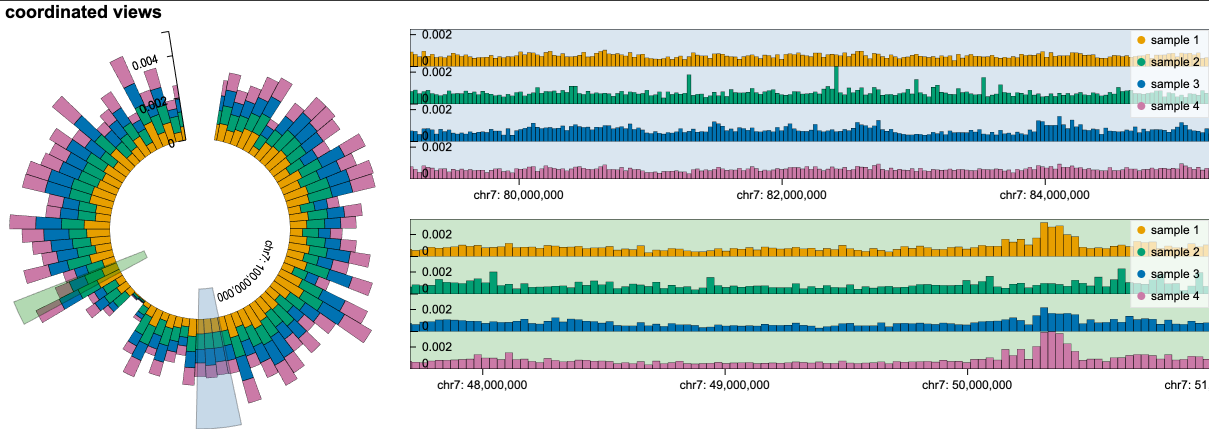
👉 Flexible View Composition#
gos supports flexible view composition.
For example, we can specify 1) the layout of a view as either "circular" or "linear" and 2) the arrangement of views as either horizontal (gos.horizontal) or vertical ("gos.vertical")
chart = gos.horizontal(
overview_2brush.properties(layout="circular", width= 400),
gos.vertical(
detail.properties(height=150),
detail2.properties(height=150),
)
).properties(title='coordinated views')
chart
gos.vertical(
overview_2brush.properties(layout="circular", width= 600, static=True),
gos.horizontal(
detail.properties(width=300).view().properties( layout="circular"),
detail2.properties(width=300).view().properties( layout="circular"),
)
).properties(title='coordinated views')
💪 Hands-on Exercise#
change the genomic domain (e.g., from
chr7tochr5) of the overview and the two detailed viewschange the multi-view arrangement (e.g., overview at the top, detailed view at the bottom)
change the encoding of the detailed views (e.g., bar charts to area charts)
Example 2: Comparative Matrix#

The two matrix visulaizations are linked to facilitate the comparision. The zooming and panning performed in one view will be automatically applied to the linked view
# Data
micro_c = gos.matrix("https://server.gosling-lang.org/api/v1/tileset_info/?d=hffc6-microc-hg38")
hi_c = gos.matrix("https://server.gosling-lang.org/api/v1/tileset_info/?d=hffc6-hic-hg38")
👉 Create a matrix visualization for the micro-c data#
size = 375
micro_c_matrix = gos.Track(micro_c).mark_rect().encode(
x=gos.X("xs:G", domain = gos.GenomicDomain(chromosome="7", interval=[77700000, 81000000])),
xe="xe:G",
y="ys:G",
ye="ye:G",
color=gos.Color("value:Q", range="warm"),
).properties(title="HFFc6_Micro-C", width=size, height=size)
micro_c_matrix.view()
👉 Create a matrix visualization for the Hi-C data#
We can create a similar matrix visualization for Hi-C data by chaning data and title.
hic_matrix = micro_c_matrix.properties(data=hi_c, title="HFFc6_Hi-C")
hic_matrix.view()
👉 Link and Compare the two matrices side by side#
We can easily link the two views by assigning them the same value for linkingId.
mat = gos.horizontal(
hic_matrix,
micro_c_matrix
)
mat
Simply putting two views together will not link them.
We need to assign them the same linkingId as shown below.
mat.properties(
title="Comparison of Micro-C and Hi-C for HFFc6 Cells",
linkingId="id"
)
👉 Compose Multiple Views#
Genomic sequene data is usually added to facilitate the analysis of genomic interaction data.
Here, we add additional views to visualize the genomic sequence data along with the matrix visualization.
# specify data
HFFc6_H3K4me3 = gos.bigwig(
url="https://s3.amazonaws.com/gosling-lang.org/data/HFFc6_H3K4me3.bigWig",
column="position",
value="peak",
binSize=8,
)
HFFc6_Atacseq = gos.bigwig(
url="https://s3.amazonaws.com/gosling-lang.org/data/HFFc6_Atacseq.mRp.clN.bigWig",
column="position",
value="peak",
binSize=8
)
HFFC6_CTCF = gos.bigwig(
url="https://s3.amazonaws.com/gosling-lang.org/data/HFFC6_CTCF.mRp.clN.bigWig",
column="position",
value="peak",
binSize=8,
)
genes = gos.beddb(
url="https://server.gosling-lang.org/api/v1/tileset_info/?d=gene-annotation",
genomicFields=[
{"index": 1, "name": "start"},
{"index": 2, "name": "end"},
],
valueFields=[
{"index": 5, "name": "strand", "type": "nominal"},
{"index": 3, "name": "name", "type": "nominal"},
],
)
epilogos_data = gos.multivec(
url="https://server.gosling-lang.org/api/v1/tileset_info/?d=epilogos-hg38",
row="category",
column="position",
value="value",
categories=[
"Active TSS", "Flanking Active TSS", "Transcr at gene 5\\' and 3\\'",
"Strong transcription", "Weak transcription", "Genic enhancers",
"Enhancers", "ZNF genes & repeats", "Heterochromatin",
"Bivalent/Poised TSS", "Flanking Bivalent TSS/Enh", "Bivalent Enhancer",
"Repressed PolyComb", "Weak Repressed PolyComb", "Quiescent/Low",
],
binSize=8,
)
# create tracks for each data resources
track_height = size / 15
track1 = gos.Track(HFFc6_H3K4me3).mark_bar().encode(
x=gos.X("start:G", axis="top"),
xe="end:G",
y=gos.Y("peak:Q", axis="none"),
color=gos.value("darkgreen"),
).properties(title="HFFc6_H3K4me3", width=size, height=track_height)
track2 = gos.Track(HFFc6_Atacseq).mark_bar().encode(
x="start:G",
xe="end:G",
y=gos.Y("peak:Q", axis="none"),
color=gos.value("#E79F00"),
).properties(title="HFFc6_ATAC", width=size, height=track_height)
gene_anno_base = gos.Track(genes).encode(
x="start:G",
size=gos.value(13),
stroke=gos.value("white"),
strokeWidth=gos.value(1),
color=gos.value("#CB7AA7"),
row=gos.Row("strand:N", domain=["+", "-"]),
)
ctcf = gos.Track(HFFC6_CTCF).mark_bar().encode(
x="start:G",
xe="end:G",
y=gos.Y("peak:Q", axis="none"),
color=gos.value("#0072B2")
)
track3 = gos.overlay(
ctcf,
gene_anno_base.mark_triangleRight(backgroundOpacity=0).encode(
color=gos.value("#CB7AA7"),
).transform_filter("strand", oneOf=["+"]),
gene_anno_base.mark_triangleLeft(backgroundOpacity=0).encode(
color=gos.value("#029F73"),
).transform_filter("strand", oneOf=["-"]).properties(title="HFFC6_CTCF")
).properties(width=size, height=track_height)
# Configure Layout
side_tracks = gos.stack(
track1.properties(height=size, width=track_height),
track2.properties(height=size, width=track_height),
track3.properties(height=size, width=track_height),
).properties(
orientation="vertical",
yOffset=size / 2.78, #
)
top_tracks = gos.stack(track1, track2, track3)
bottom_tracks = gos.Track(epilogos_data).mark_bar().encode(
x=gos.X("start:G", axis="none"),
xe="end:G",
y=gos.Y("value:Q", axis="none"),
color=gos.Color("category:N", range=[
"#FF0000", "#FF4500", "#32CD32", "#008000", "#006400",
"#C2E105", "#FFFF00", "#66CDAA", "#8A91D0", "#CD5C5C",
"#E9967A", "#BDB76B", "#808080", "#C0C0C0", "gray"
]),
).transform_filter("value", inRange=[0, 999999]).properties(
title="Epilogos (hg38)",
width=size,
height=track_height,
).view()
Let’s see how the three additional views look like
bottom_tracks
Compose and link multive views
We use a same linkingId to link all views in this visualization.
left_view = gos.vertical(top_tracks, hic_matrix, bottom_tracks, spacing=0)
right_view = gos.vertical(
top_tracks,
# replace data source for right side, change title
micro_c_matrix,
bottom_tracks,
spacing=0,
)
main= gos.horizontal(left_view, right_view, spacing=30)
comp_matrix_vis = gos.horizontal(side_tracks, main).properties(
title="Matrix Visualization",
subtitle="Comparison of Hi-C and Micro-C for HFFc6 Cells",
xDomain=gos.GenomicDomain(chromosome="7", interval=[77700000, 81000000]),
spacing=1,
linkingId="-"
)
comp_matrix_vis
In the above visualization, multiple views (top_tracks, bottom_tracks, side_tracks, hic_matrix, micro_c_matrix) are oragnized as below:
gos.horizontal/
├─ side_tracks
└─ gos.horizontal/
├─ gos.vertical/
│ ├── top_tracks
│ ├── hic_matrix
│ └── bottom_tracks
└─ gos.vertical/
├── top_tracks
├── micro_c_matrix
└── bottom_tracks
💪 Hands-on Exercises#
Modify the arrangements of the
top_tracks,side_tracks,bottom_tracks, and the two matrices.
Save and Load Gosling Visualizations using JSON Spec#
comp_matrix_vis.to_json()
with open('gos_comp_mat_spec.json', 'w') as f:
json.dump(comp_matrix_vis.to_dict(), f)
files.download('gos_comp_mat_spec.json')
with open('gos_comp_mat_spec.json', 'r') as f:
spec = json.load(f)
gos.View(**spec)
gos.View(**mat.to_dict())
with open('mat_spec.json', 'w') as f:
json.dump(mat.to_dict(), f)
files.download('mat_spec.json')